Alumni Ferrari

{kind=link}
DNA DAMAGE AND THE CELL CYCLE
Introduction
The increased survival of cancer patients treated with aggressive radio- or chemotherapy comes at a price, with secondary leukemia being one of the most common causes of death. In recent years, the rational design of molecules targeting oncogenic pathways that are hyper-functional in cancer cells or to which cancer cells become addicted has provided an alternative and promising approach to the use of “dirty” drugs. The established adaptability of cancer cells to external insults, however, constantly demands the implementation of novel strategies. Basic research aims to satisfy this demand through the continuous discovery of novel targets.
On one front, we are investigating the cellular response to genotoxic stress, to elucidate the impact of DNA damage checkpoint activation on the onset of mitosis.
On another front, we are engaged in the identification and characterization of small molecule inhibitors of mitotic ATPases and dual-specificity phosphatases.
Mitotic ATPases and genome stability
Inappropriate detection or untimely repair of DNA damage before the onset of mitosis may lead to chromosome breaks as well as rearrangements or fusions – comprehensively know as “structural abnormalities” - that lead to the development of cancer and have been focus of intense research in the last decades. On the other hand, loss or gain of entire chromosomes – known as “numerical abnormalities” - is a characteristic of tumors described more than a century ago and named “aneuploidy”. The most common causes of aneuploidy are: (i) defective attachment of sister chromatids by spindle microtubules (merotelic attachment) in pro-metaphase and metaphase, often linked to centrosome amplification, (ii) malfunction of the spindle assembly checkpoint; (iii) defects in chromosome cohesion.
RUVBL1 and RUVBL2 are essential genes belonging to the AAA+ superfamily (ATPases Associated with various cellular Activities). The two polypeptides form homo- and hetero-hexamers and as such are part of large multiprotein complexes involved in transcription, chromatin remodeling and DNA mismatch repair. Interestingly, expression of both RUVBL1 and RUVBL2 is upregulated in cancer.
In this study we addressed the mitotic role of RUVBL1. We observed that at anaphase-to-telophase transition, RUVBL1 localizes to structures of the mitotic spindle apparatus, partially co-localizing with polo-like kinase 1 (PLK1). Interestingly, RUVBL1 and RUVBL2 separate during cytokinesis, with only RUVBL1 remaining co-localized with PLK1. PLK1 is able to phosphorylate RUVBL1 - but not RUVBL2 - at a single site (T239). This, along with the evidence of physical association between RUVBL1 and PLK1 in vivo,suggests that the latter may differentially regulate the function of the RuvB-like proteins during mitosis. Regarding the function of RUVBL1/2 in mitosis, we observed that depletion of RUVBL1 or RUVBL2 expression by RNAi causes extension of metaphase and retardation of the anaphase onset, with polar chromosomes failing to align at the metaphase plate and ultimately lagging at anaphase. Finally, we could show that the ATPase activity of RUVBL1 is indispensable for cell proliferation.
Current studies aim at elucidating the role of RUVBL1 in the molecular mechanism that facilitates polar chromosome congression at metaphase and the identification of RUVBL1 small molecule inhibitors.
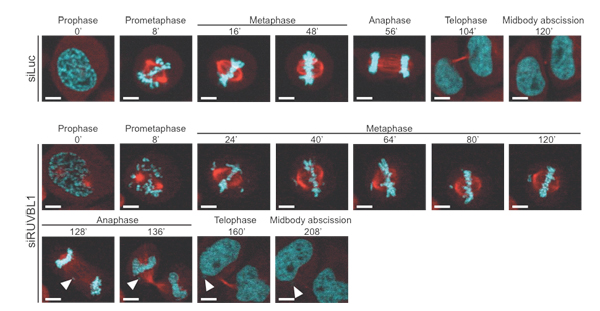
Figure 1.
RUVBL1 depletion affects the length of mitosis and
results in lagging chromosomes.
RNAi-mediated
knock-down studies followed by confocal live imaging of HeLa cells. Stills of
control (Luc) or RUVBL1 siRNA-treated cells are shown. Lagging chromosomes are
indicated with arrowheads. DNA is shown in cyan and α-tubulin in red.
Control of Exo1 by phosphorylation
Double-strand breaks (DSBs) are among the most cytotoxic DNA lesions generated by ionizing radiation (IR), certain chemotherapeutic drugs, collapse of stalled DNA replication forks or during physiological processes such as meiotic recombination. DSBs repair occurs through error-prone (Non-Homologous-End-Joining) or error-free (Homologous-Directed-Repair) pathways. Rapid and reversible control of pathways components is exerted through post-translational modifications (PTMs). To shed light on the role of PTMs that control DNA end-resection guiding error-free repair of DSBs, we focused on a key factor of HDR pathways, the 5’-to-3’ exonuclease-1 (EXO1). EXO1 is a Rad2 family DNA repair nuclease originally identified in S. pombe and shown to participate in several post-replication DNA repair pathways including mismatch repair, DSBs repair, meiotic and mitotic recombination. Additionally, yeast Exo1 was shown to localize at stalled replication forks where it counteracts fork reversal.
Studies that we conducted in the past showed that human EXO1 is regulated by sumoylation, which likely facilitates its recruitment at stalled DNA replication forks where it undergoes phosphorylation-dependent poly-ubiquitylation, which results in its proteasome-mediated degradation, thus avoiding pathological resection of DNA. At DSBs, human EXO1 functionally interacts with CtIP/RBBP8 that modulates both EXO1 recruitment at DNA ends and its resection activity. On the other hand, we showed that yeast Exo1 is regulated through interaction with 14-3-3 proteins and others provided evidence for inhibition of Exo1 activity by phosphorylation.
In this study we identified kinase and phosphatase controlling Exo1 phosphorylation upon stalled DNA replication as well as the entire complement of checkpoint-dependent phosphorylation sites in Exo1. Functional studies conducted in exo1Δ sgs1Δ strains and addressing the role of phosphorylation on Exo1 resection activity at DSBs are ongoing.
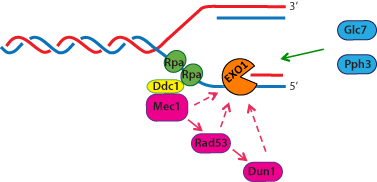
Figure 2.
Phosphorylation-dependent control of yeast Exo1 at stalled replication forks.
Upon
stalled DNA replication, Exo1
is phosphorylated in a checkpoint-dependent manner at five major sites. During checkpoint
recovery, Exo1 dephosphorylation is likely controlled by the protein
phosphatases Glc7 and Pph3.
CDC25 inhibitors
The network of signals originating at sites of damage and triggering cell cycle checkpoints involves the action of protein kinases and protein phosphatases. The protein phosphatase CDC25 is an oncogene overexpressed in a number of cancers that controls activation of Cyclin-Dependent Kinases (CDKs) at key cell cycle transitions. Aim of this study is the identification and characterization of small molecule CDC25 inhibitors. To this end, we first identified a common pharmacophore (i.e., a set of structural features necessary for molecular recognition of a ligand by a macromolecule) through in silico fragmentation of all known CDC25 inhibitors. Next, we conducted a similarity search of a molecule databases (>2000 structures) with the obtained fragments and finally we performed molecular docking of selected scaffolds into the catalytic pocket of CDC25, the crystal structure of which is available. Enzymatic assays followed by cellular studies led to the identification of 8 scaffolds that are undergoing modeling-guided synthesis to provide second-generation compounds that will be profiled in enzymatic assays and tested in 2D and 3D cell culture systems for their efficacy as CDC25 inhibitors.
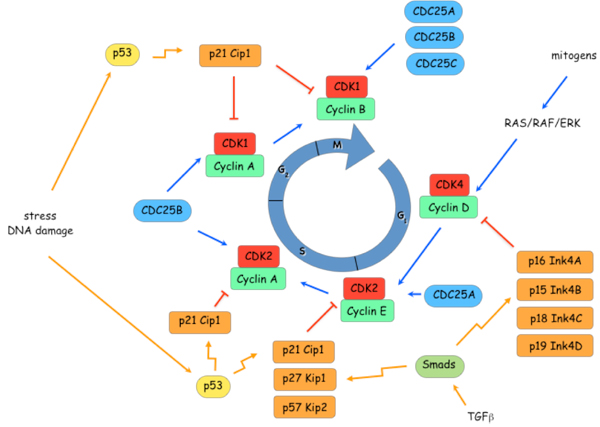
Figure 3. Control of cell cycle transitions.
The point of action of
different Cyclin-CDK complexes during transition through the cell cycle and the
network of positive and negative inputs are shown.
Photo-activatable metal-based complexes and cancer therapy
Photodynamic therapy (PDT) is an attractive alternative to chemotherapy. Among the different photosensitizers (PSs) employed, Ru(II) polypyridyl complexes were found to be valid substitutes to porphyrin-based or phtalocyanine-based PSs. In this study we obtained evidence that one such complex, namely [Ru(bipy)2-dppz-7-methoxy][PF6]2 (Ru65), localized in the nucleus of various cancer and normal cells and displayed cytotoxicity only upon UV-A irradiation. Importantly, we demonstrated that Ru65 intercalated in DNA and, upon light irradiation, promoted guanine oxidation, resulting in nicks in the double helix. We confirmed this mechanism of action in living cells showing that UV-A irradiation of cells loaded with Ru65 resulted in a transient DNA damage response. Strikingly, photo-irradiation of Ru65 triggered cell death upon resolution of DNA damage and with distinct mechanisms in interphase or mitotic cells. Interphase cells underwent cell cycle arrest at the G2/M phase and massive cytoplasmic vacuolation, which was paralleled by an unfolded-protein stress-response, resulting in reduction of viability and cell death through a paraptosis-like mechanism. On the other hand, UV-A irradiation of Ru65 in G2/M synchronized cells did not allow completion of mitosis and rapid cell death through classic apoptotic pathways ensued. Importantly, targeting mitotic cells with Ru65 allowed to significantly increase its photo-toxicity. Overall, our findings allow to draw two important conclusions: (i) PSs targeting the nucleus and causing a transient DNA damage response upon photoactivation represent an excellent alternative to current chemotherapeutics; (ii) combination of cell cycle inhibitors and PSs appears to be an even better alternative to the use of PSs alone for an effective clearance of cancer cells.
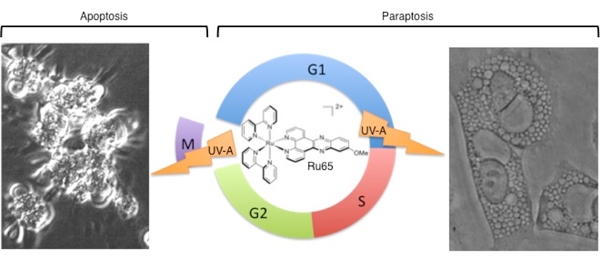
Figure 4.
Dual mode of cell
death upon photo-irradiation of
Ru65 in interphase and mitosis.
Scheme of the different cell death modes triggered
by UV-A irradiation of Ru65 in interphase or at mitosis.