Research
DNA damage and repair: From molecular mechanisms to targeted therapy
Studying the causes and consequences of DNA damage on the molecular and cellular level is a major topic of research within cancer biology. DNA damage not only causes cancer but is also used as a means to treat cancers through radio- or chemotherapy and responsible for their unwanted side effects. Therefore, a better understanding of the underlying mechanisms activated in response to DNA damage is of utmost importance for the development of novel anti-cancer therapies.
Background
The main focus of our laboratory is to gain molecular and genetic insights into the interplay between DNA damage signaling and repair in human cells. Within this broad area of research, we are particularly interested in the repair of DNA double-strand breaks (DSBs), the most hazardous lesions a cell can encounter. A detailed knowledge of the players acting in DSB repair processes is crucial if we are to improve current cancer therapies and suggest novel strategies to fight this disease.
DSBs are repaired by two mechanisms: homologous recombination (HR) and non-homologous end-joining (NHEJ). The human CtIP protein plays a critical role in DSB repair pathway choice by promoting DNA-end resection and HR (Figure 1). We are employing several approaches to further our understanding on the function and regulation of CtIP and other key DSB repair proteins by multiple mechanisms, involving cell cycle control, post-translational modifications (PTMs) and protein-protein interactions (PPIs). Besides the exciting prospect of identifying novel factors involved in DSB repair by high-throughput proteomic screens, we use a combination of biochemistry, cell biology and genetics to address the function of CtIP and other factors implicated in the maintenance of genome stability (Figure 2).
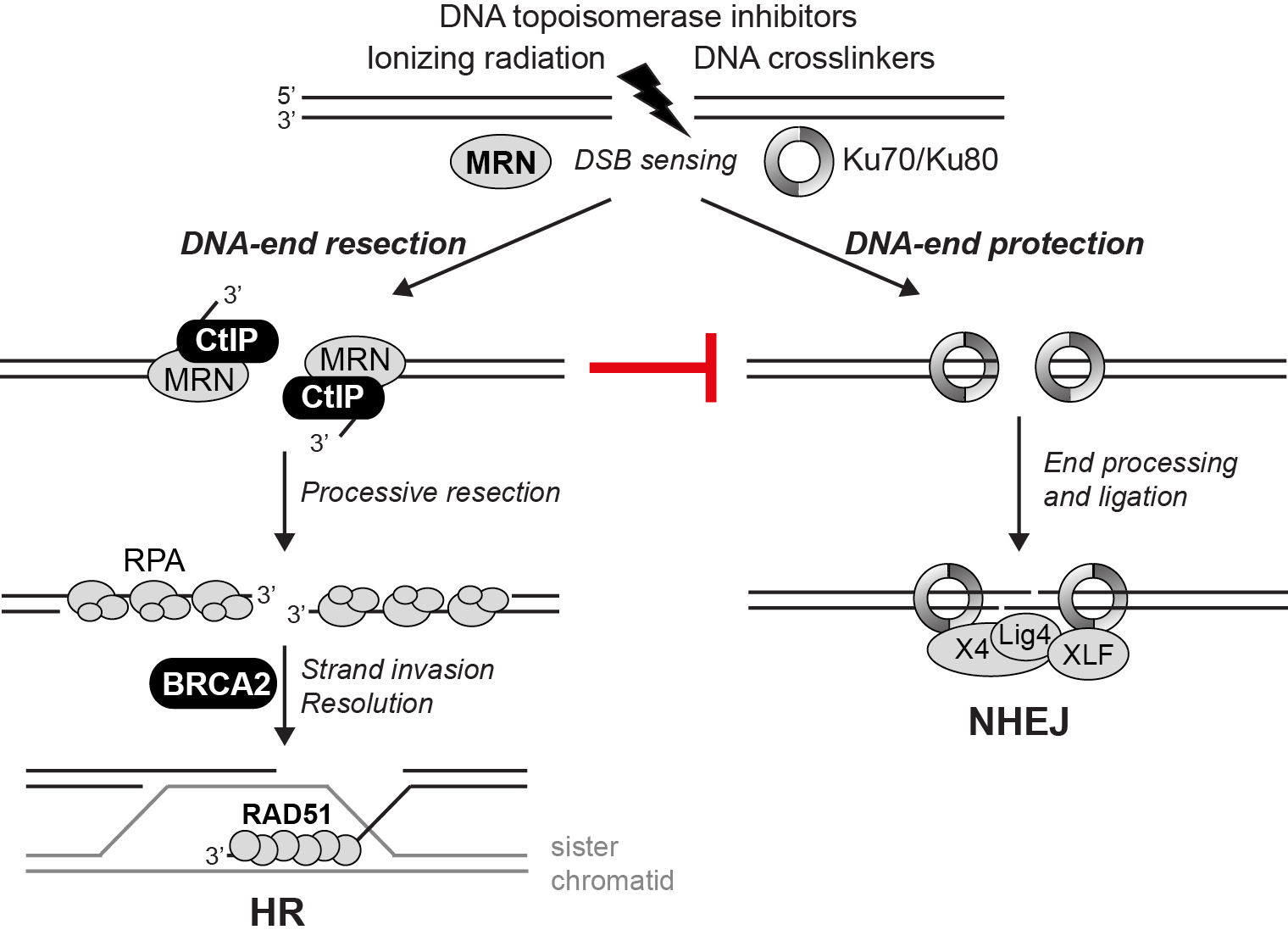
Figure 1. Mechanistic model depicting the role of CtIP in DSB repair pathway choice. DSBs induced by a variety of exogenous agents used in cancer therapy are sensed by either the MRN complex or the Ku heterodimer. CtIP and MRN are essential for initiating DNA-end resection, the first step in HR, and suppressing NHEJ. In HR, initial processing of DSBs is followed by processive resection through the combined action of DNA helicases and nucleases. The 3'-single-stranded DNA (ssDNA) is protected from degradation through the binding of RPA. BRCA2 promotes replacement of RPA by the RAD51 recombinase, which is required for strand invasion of the sister chromatid promoting D-loop formation and further downstream steps in HR.
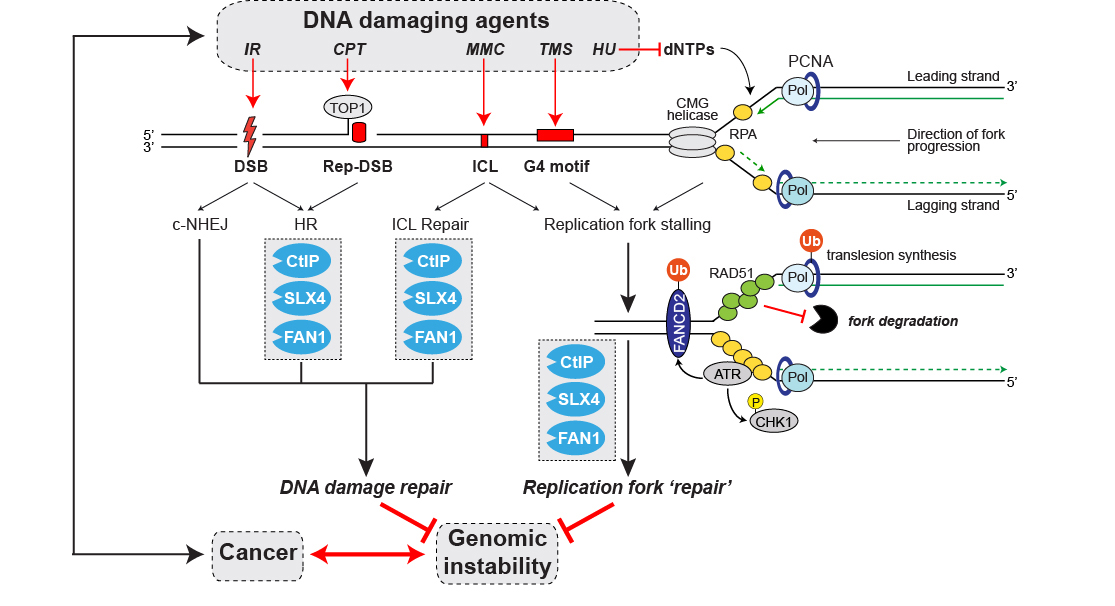
Figure 2. CtIP, FAN1 and SLX4 DNA repair nucleases: Caretakers required for the maintenance of genome integrity and cancer avoidance. Schematic illustration highlighting the roles of CtIP, SLX4 and FAN1 proteins in response to a variety of DNA damaging agents causing different types of DNA lesions. Abbreviations: IR, ionizing radiation; CPT, camptothecin; MMC; mitomycin C; TMS, telomestatin; HU, hydroxyurea; DSB, double-strand break; Rep-DSB, replication-dependent DSB; ICL, DNA interstrand crosslink; G4, guanine-rich DNA sequence motifs potentially forming four-stranded G-quadruplex structures during DNA replication; CMG, Cdc45/Mcm2-7/GINS; Pol, DNA polymerase; c-NHEJ, classical non-homologous end-joining; HR; homologous recombination; Ub, ubiquitin; P, phospho.
Ongoing Projects
The role of PIN1 isomerase in the maintenance of genome stability
An accurate regulation of the DNA damage response (DDR) by phosphorylation-dependent signalling is absolutely pivotal to trigger correct repair of the DNA. Inappropriate regulation can have detrimental effects for the cell fate, hence incorrect repair can cause tumorigenesis. However, surprisingly little is known about the DDR phosphorylation-network and its fine-tuning. Human PIN1 (peptidyl-prolyl cis-trans isomerase NIMA-interacting 1) specifically recognizes and isomerizes phosphorylated serine/threonine-proline (pS/TP) motifs, thereby inducing a cis-trans conformational change in the target protein. This signalling mechanism has profound consequences on protein function and stability.
Our lab has reported that PIN1 regulates DSB repair pathway choice via CtIP isomerization (Steger et al., Mol Cell. 2013). Moreover, using a proteomic screen, we discovered several key DDR factors as potential PIN1 substrates, including BRCA1, MDC1 and SLX4. SLX4 is a multi-domain scaffold protein for several structure-specific nucleases involved in the cleavage of DNA interstrand-crosslinks (ICLs) and the resolution of Holliday junctions during HR. Our current data firmly establishes that PIN1 binds to SLX4 in a phosphorylation-dependent manner. In this ongoing project, we aim to functionally characterize the role of site-specific SLX4 phosphorylation and subsequent isomerization in the DDR, further highlighting the importance of PIN1 in the maintenance of genome stability.
Separating the function of human CtIP at DNA breaks and replication forks
Faithful genome duplication is a major and challenging task of every cell in our body. However, replication forks frequently encounter obstacles that impede their progression leading to fork stalling, a condition collectively referred to as 'DNA replication stress'. Replication stress can arise from many intrinsic and extrinsic sources, such as secondary DNA structures, collisions between replication and transcription complexes, repetitive sequences, irradiation or genotoxic agents. Strikingly, unresolved replication stress leads to genomic instability, a frequent hallmark of carcinogenesis. Over the past decade, proteins involved in homologous recombination (HR) pathway have been identified to play a separate and crucial role in stabilizing and restarting stalled forks. For instance, BRCA1 and BRCA2 tumour suppressor proteins were shown to protect replication forks from MRE11-dependent degradation, thereby counteracting fork collapse. In addition, our lab has recently demonstrated that CtIP protects reversed forks from degradation by the DNA2 nuclease (Przetocka et al., Mol Cell. 2018) (Figure 3). We now aim to genetically dissect the underlying mechanisms governing CtIP’s function in HR versus fork protection.
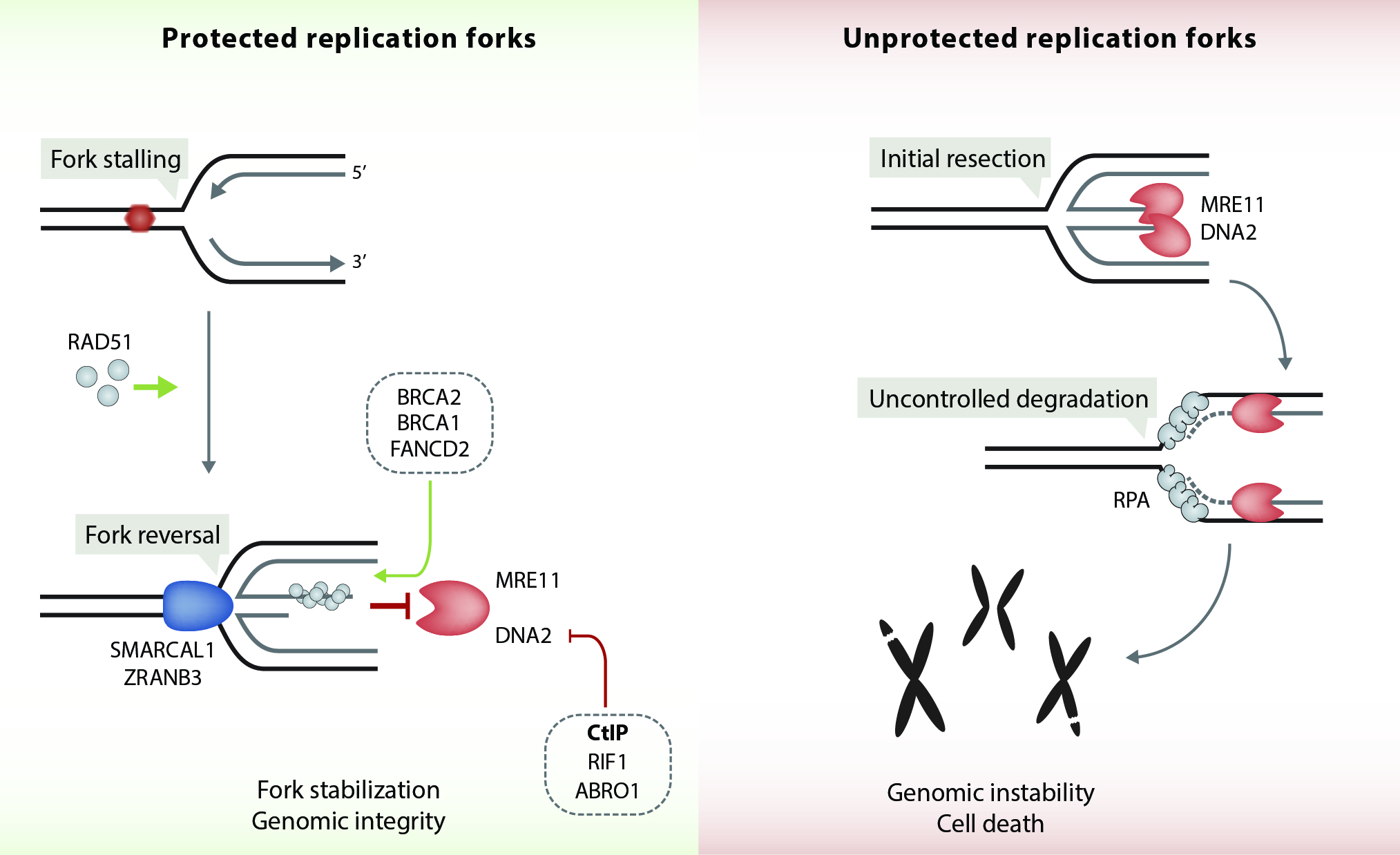
Figure 3. CtIP protects stalled replication forks from DNA2-mediated degradation. RAD51 together with DNA translocases (SMARCAL1, ZRANB3) mediate the reversal of stalled replication forks. RAD51-ssDNA filament formation and stabilization at reversed forks is facilitated by BRCA1, BRCA2 and FANCD2. Together, these factors prevent MRE11 exonuclease from progressively degrading nascent DNA at reversed forks. In addition, CtIP, ABRO1 and RIF1 protect reversed fork from DNA2-mediated degradation. In the absence of fork protection factors, excessive nucleolytic processing of stalled forks causes fork collapse, leading to DSBs and genomic instability.
Towards the development of inhibitors targeting DNA double-strand break repair
Cells rely on efficient mechanisms sensing and repairing different types of DNA damage to survive and proliferate. Due to oncogene-induced replication stress and acquired defects in the DNA damage response (DDR), hyperproliferating cancer cells are becoming increasingly addicted to DSB repair mechanisms to survive genotoxic stress. Conventional radio- and chemotherapy as well as new molecular targeted therapies, primarily the use of PARP inhibitors (PARPi) in HR-deficient cancers, take advantage of the cytotoxic properties of DSBs. On the other hand, augmented or restored DSB repair capability is a major cause of treatment resistance and cancer relapse. Thus, potent DSB repair inhibitors would offer multiple therapeutic opportunities. HR relies on CtIP-dependent DNA end resection and subsequent BRCA2-dependent loading of RAD51 on ssDNA. Importantly, repair functions of BRCA2 and CtIP mostly depend on protein-protein interactions (PPIs) rather than on enzymatic activity.
Inhibition of PPIs has proven challenging as PPI interfaces commonly do not support binding of small drug-like molecules. In contrast, peptide-based inhibitors of PPIs are considered more promising, but their therapeutic use is often limited by conformational and proteolytic instability as well as low membrane permeability. Importantly, chemical modifications (unnatural amino acids, stapling or cyclization) and the use of cell-penetrating peptides (CPPs) as delivery vectors were shown to significantly improve pharmaceutical properties of peptides. In this project, we apply structured-based design of peptides as well as unbiased screening of compound libraries to identify CtIP-targeting agents (Figure 4). As a proof of principle, we have developed a native BRCA2-mimetic peptide interfering with RAD51 function in HR and fork protection (Trenner et al., Mol Cancer Ther. 2018). We anticipate that such molecules could not only be applied for basic research purposes, but may also improve the efficacy of current standard cancer therapies and potentially be used as single drugs in personalized medicine applications.
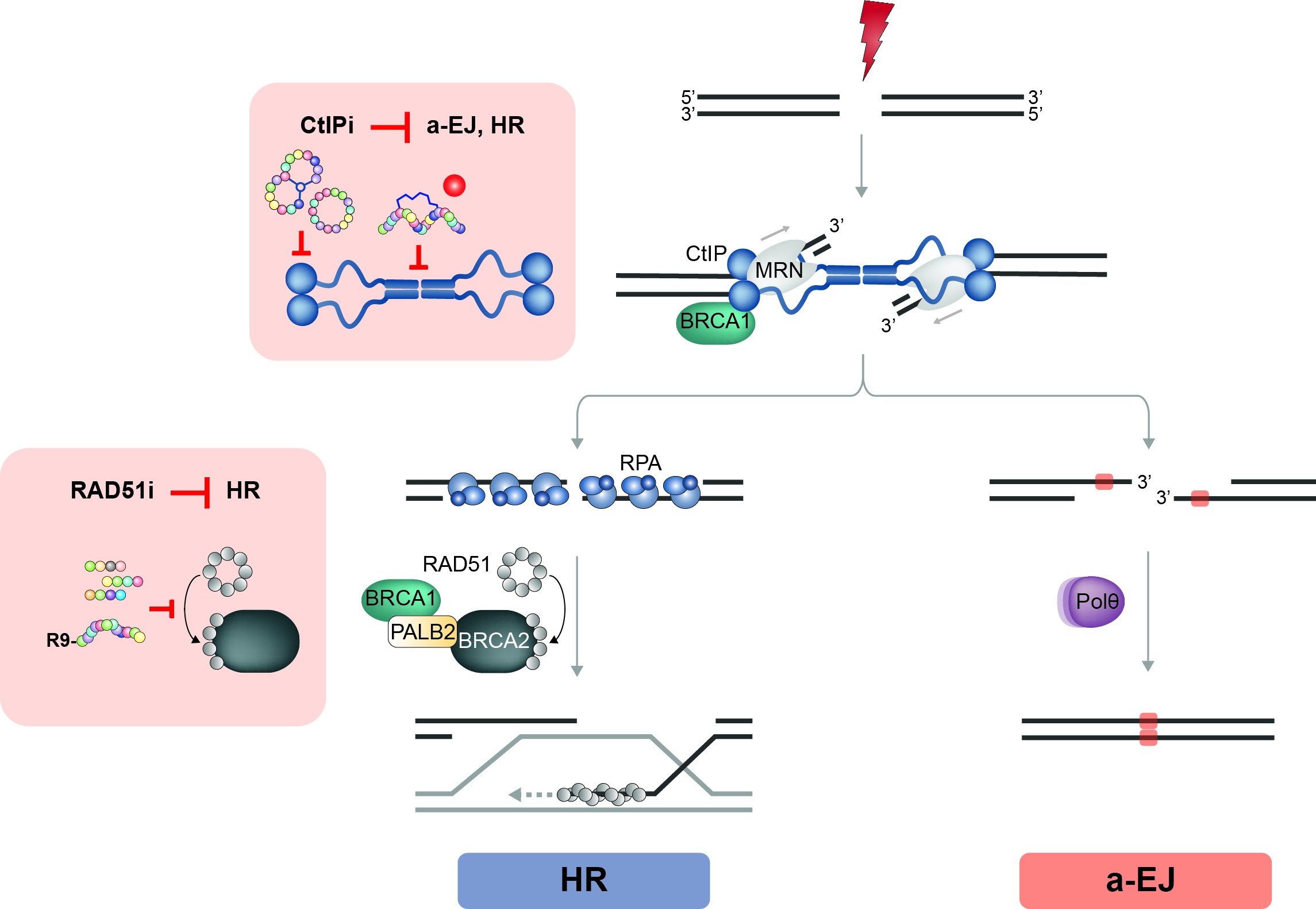
Figure 4. Designing new DSB repair targeting drugs.
DNA end resection relies on CtIP activity and is the starting point for HR and alternative end-joining (a-EJ). Consequently, whereas CtIP inhibitors (CtIPi) target both DSB repair pathways, RAD51 inhibitors (RAD51i) might specifically affect HR. Specifically, we aim to apply peptide stapling to mimic the CtIP tetramerization domain and various screening technologies (Phage Display, SICLOPPS) to generate specific peptide inhibitors of CtIP. In addition, we are in the process to identify small molecule binders of CtIP protein domains (red dots) by screening DNA-encoded chemical libraries (DECLs).
Human FAN1: Molecular insights into a multifunctional DNA repair nuclease
FANCD2-associated nuclease 1 (FAN1) is an evolutionarily conserved DNA endo- and exonuclease originally characterized as a crucial ICL processing factor, and, therefore, considered as a member of the Fanconi Anemia (FA) pathway. However, subsequent genetic studies in mice and clinical data suggest that FAN1 is not an FA gene. Instead, FAN1 mutations have been linked to karyomegalic interstitial nephritis (KIN) and cancer predisposition. Remarkably, recent genome-wide association studies (GWASs) have identified FAN1 variants that modulate progression of Huntington’s disease (HD), an inherited neurodegenerative disorder caused by an expanded CAG repeat in the huntingtin gene. FAN1’s mode of action in reducing trinucleotide repeat expansions is currently unknown. Of note, FAN1 was repeatedly reported to physically interact with MLH1, best known for its role in post-replicative mismatch repair (MMR). Interestingly, MLH1 and other MMR proteins have also been implicated in ICL processing and identified as genetic modifiers of HD, meanwhile FAN1 was found to interact with PCNA and to alleviate replication-stress induced genome instability (Porro et al., Nat Commun. 2017). Thus, in one of our ongoing FAN1 projects, we aim to define the molecular basis of the FAN1-MLH1 interaction and its biological relevance (Figure 5).
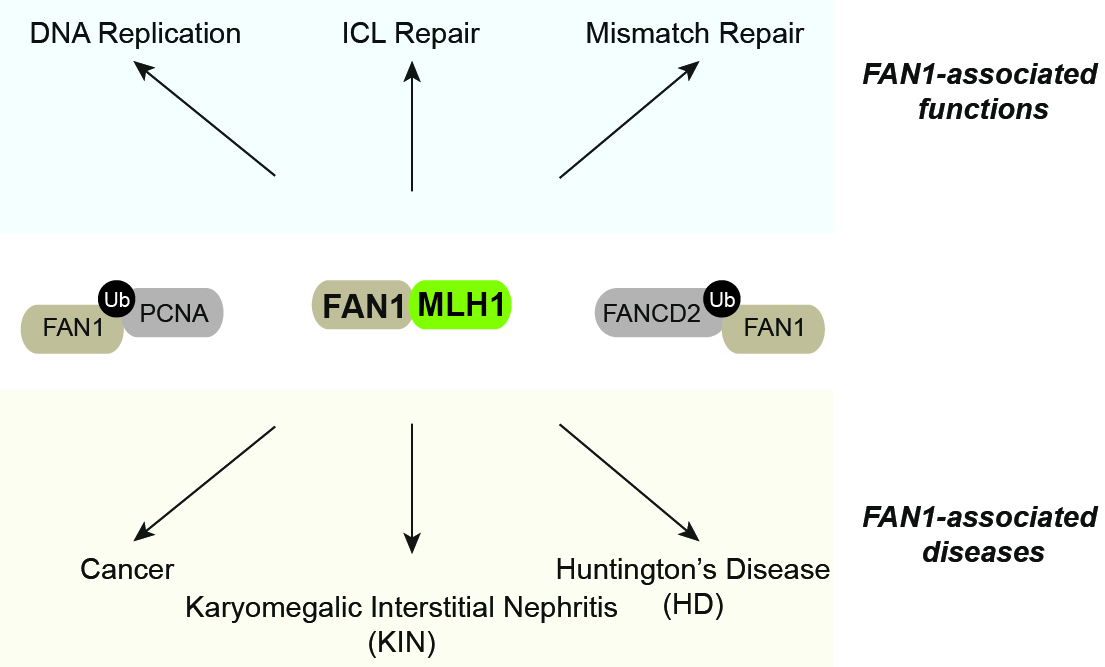
Figure 5. FAN1-associated functions and diseases. See text for details.
Our research is supported by grants from the Swiss National Science Foundation (SNSF), the Swiss Cancer Research Foundation (Krebsforschung Schweiz) and the Forschungskredit of the University of Zurich.