
Mechanisms of replication stress-induced tumorigenesis
Background
Replication stress has emerged as an early causative event in tumorigenesis, frequently detectable already in precancerous lesions and promoted by activation of well-known oncogenes. Moreover, replication stress creates selective pressure for further mutations that occasionally inactivate checkpoint control, leading to more aggressive cancers. Several tumour suppressors, originally known as DNA repair factors, were recently found to play central roles at stalled replication forks - genetically and mechanistically distinct from their function in DNA damage repair. Uncovering how oncogenes and tumour suppressors specifically operate upon endogenous and exogenous replication stress is of key relevance to obtain full mechanistic understanding of cancer onset, especially in individuals carrying known mutations predisposing to cancer.
Goal
The goal of this research line is to use our portfolio of imaging and molecular approaches to investigate the role of known oncogenes and tumour suppressors in the replication stress response. We aim to uncover how specific oncogenes exacerbate endogenous sources of genome instability and/or affect the cellular responses to these challenges. Similarly, we attempt to identify novel roles of known tumour suppressors in the replication stress response and to characterize how genetic alterations associated with increased cancer susceptibility impact on genome integrity during replication.
Ongoing and future work
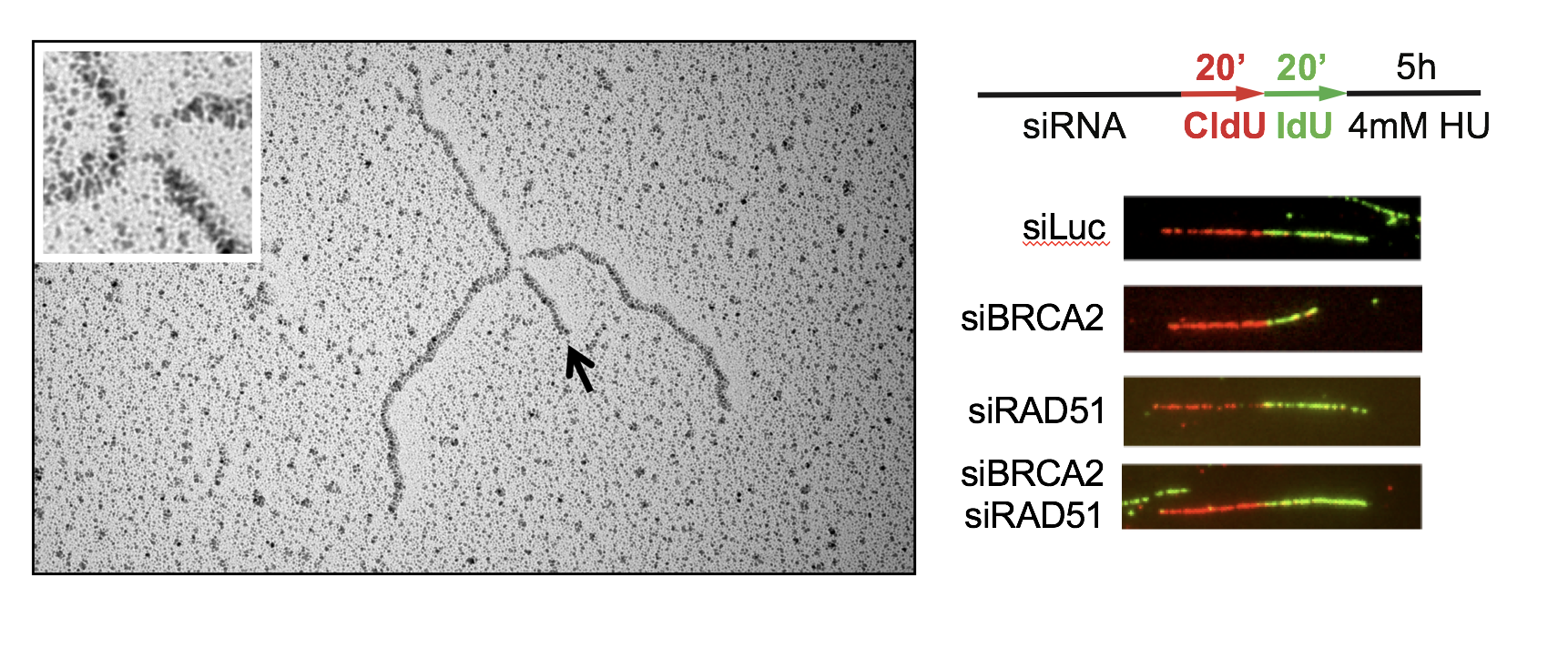
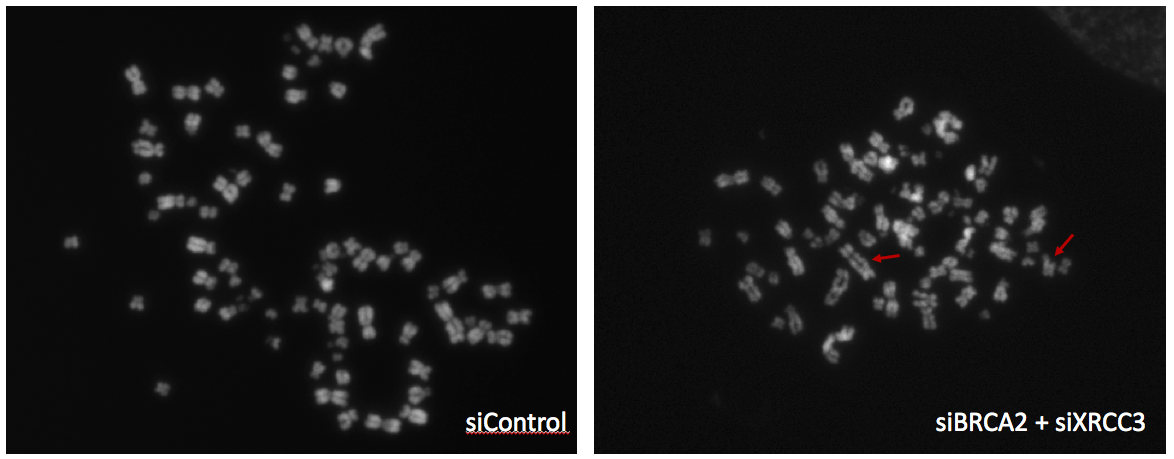
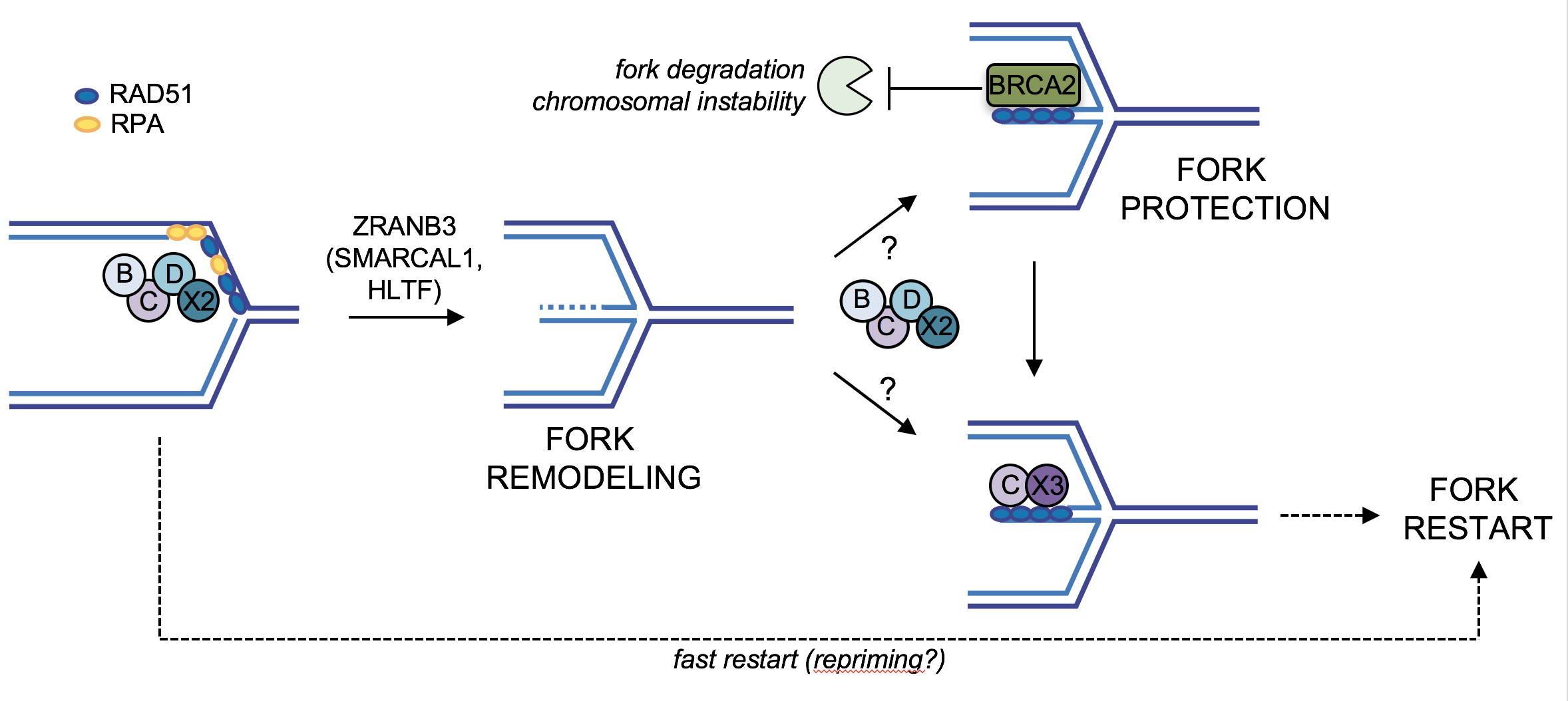
Our work in this area uncovered replication fork remodelling as common and rapid consequence of the activation of different oncogenes (Neelsen et al., J. Cell Biol 2013). Moreover, we identified an important role in fork remodelling and protection of several tumour suppressors, well known in the context of homologous recombination repair (BRCA2, RAD51, RAD51 paralogs, etc.). As oncogenes frequently alter the transcriptional program while boosting proliferation and replication, we recently investigated key molecular mechanisms occurring at transcription-replication conflicts (TRCs), often associated with RNA-DNA hybrid accumulation. To reach this goal, we established several approaches to monitor key intermediates associated with TRCs and we now plan to exploit this experimental platform to investigate whether and how TRCs underlie genomic instability and tumorigenesis in specific cell types, particularly exposed to this peculiar type of replication stress. We also aim to clarify whether in these cells even partial inactivation of certain tumour suppressors may lead to increased genomic instability, contributing to the associated cancer susceptibility.
Recent selected publications
M. Vujanovic, J. Krietsch, M.C. Raso, N. Terraneo, R. Zellweger, J.A. Schmid, A. Taglialatela, J.W. Huang, C.L. Holland, K. Zwicky, R. Herrador, H. Jacobs, D. Cortez, A. Ciccia, L. Penengo and M. Lopes (2017). Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Molecular Cell, 67:882-890.
S. Mijic, R. Zellweger, N. Chappidi, M. Berti, K. Jacobs, K. Mutreja, S. Ursich, A. Ray Chaudhuri, A. Nussenzweig, P. Janscak and M. Lopes (2017). Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nature Communications, 8(1):859.
M. Berti, F. Teloni, S. Mijic, S. Ursich, J. Fuchs, M. D. Palumbieri, J. Krietsch, J. A. Schmid, E. B. Garcin, S. Gon, M. Modesti, M. Altmeyer, and M. Lopes (2020). Sequential role of RAD51 paralog complexes in replication fork remodeling and restart. Nature Communications, 11(1):3531.
Direct R-Loop Visualization on Genomic DNA by Native Automated Electron Microscopy. Stoy H, Luethi J, Roessler FK, Riemann J, Kaech A, Lopes M.Methods Mol Biol. 2022;2528:1-20. doi: 10.1007/978-1-0716-2477-7_1.
H. Stoy, K. Zwicky, D. Kuster, K.S. Lang, J. Krietsch, M. P. Crossley, J. A. Schmid, K. A. Cimprich, H. Merrikh and M. Lopes (2023). Direct visualization of transcription-replication conflicts reveals post-replicative DNA:RNA hybrids. Nature Struct Mol Biol, doi: 10.1038/s41594-023-00928-6